过程工程所建立国际上首个GPU加速的化学反应分子动力学程序GMD-Reax
自2007年以来,将原来专门用于图像处理的GPU(Graphics Processing Unit)卡应用于高性能科学计算、特别是分子动力学计算的尝试成为热点,国际上相关的进展日新月异,利用GPU并行提升经典MD模拟时空尺度的能力、特别是在桌面计算机上进行大规模体系MD模拟的前景逐步明朗,但经典MD模拟的主要是分子体系的物理过程,不能应用于含化学反应的过程。量子力学(化学)方法是目前能够从分子水平计算化学反应的主要方法,但因其需要预先假定反应路径、且计算代价高昂,单一计算节点可应用的规模在数百个原子(~100量级)的简单体系;Van Duin等提出并正在完善的化学反应力场ReaxFF与经典MD结合可将单节点计算化学反应的能力提高到数千个原子(~1,000量级)。如果利用GPU加速ReaxFF MD,则可将计算化学反应体系的能力至少提高到数万个原子(~10,000量级)甚至更多,从而使一些大规模体系的复杂化学反应的模拟如煤热解的模拟成为可能。
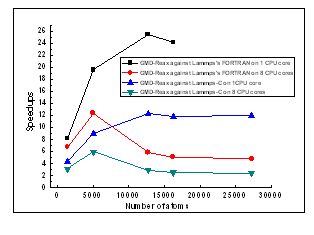
高性能计算与化学信息学课题组在国际上建立了首个基于单GPU加速的化学反应分子动力学程序GMD-Reax,其单节点计算能力(相比于8核CPU)比国际知名的LAMMPS平台(http://lammps.sandia.gov/)上的相应程序提升数倍。GMD-Reax的算法文章已发表在美国化学会计算机化学分会的期刊Journal of Molecular Graphics and Modelling (Mo Zheng, Xiaoxia Li, Li Guo, Algorithms of GPU-enabled reactive force field (ReaxFF) molecular dynamics. Journal of Molecular Graphics and Modelling 2013, 41, (April), 1-11),确立了思路和程序性能的先进性。ReaxFF方法的作者van Duin教授、以及该方法LAMMPS平台CPU并行程序的作者Aktulga博士均已来信交流。
(高性能计算与化学信息学课题组 李晓霞)